Leveraging RDKit, PyTorch, and TensorFlow in Molecular Design

In the realm of computational chemistry, the integration of advanced machine learning frameworks with chemical informatics tools has opened new avenues for molecular design. Here, we explore how RDKit, alongside deep learning libraries like PyTorch and TensorFlow, and the more recent MatterGen, are being utilized to innovate in this space:
RDKit and Molecular Representation:
- RDKit serves as a primary tool for molecule manipulation and representation. It offers functionalities to convert SMILES strings into molecular graphs, which is foundational for any AI-driven molecular design. The Oxford Protein Informatics Group provides insights on transforming SMILES into graph structures suitable for Graph Neural Networks (GNNs) using RDKit, thus facilitating the application of machine learning models on chemical data (1, 6).
PyTorch for Dynamic Molecular Modeling:
- PyTorch has gained traction for its dynamic computation graphs, which are particularly beneficial for molecule design where the model's architecture might need to be adjusted based on the chemical complexity or task at hand. A notable application is in generating molecular graphs for tasks like solubility prediction or drug discovery. Blog posts by iwatobipen delve into using PyTorch for building Graph Convolutional Networks (GCNs) for Quantitative Structure-Activity Relationship (QSAR) models, demonstrating PyTorch's flexibility in handling molecular data (3).
TensorFlow for Scalable Molecular Design:
- TensorFlow's strength lies in its scalability and ease of deployment, making it suitable for large-scale molecular simulations and predictions. While TensorFlow's static graph model might be less flexible than PyTorch's for rapid prototyping, it excels in distributed computing environments. The blog "Cheminformania" by Esben Jannik Bjerrum discusses using TensorFlow for reaction prediction, showcasing how TensorFlow can manage complex chemical transformations (4).
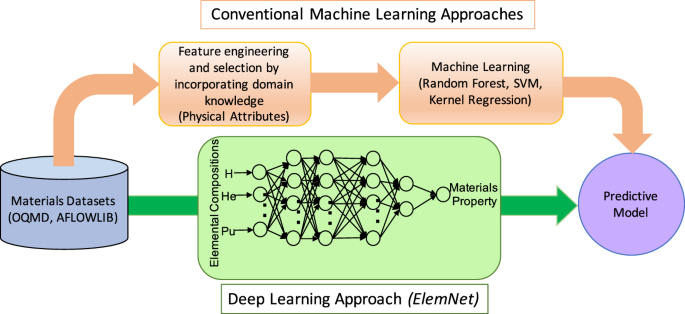
My Comparative Insights:
- Both frameworks, PyTorch and TensorFlow, have their merits. PyTorch is often praised for its research-friendly environment, allowing for quick experimentation and debugging, which is crucial in the iterative process of molecule design. On the other hand, TensorFlow is favored for its robustness in production environments, particularly for deploying models at scale. Discussions on platforms like Real Python and Stackify highlight these differences, guiding practitioners in choosing the right tool for their specific needs.
Integration and Future Directions:
- The integration of RDKit with either PyTorch or TensorFlow allows for a comprehensive approach to molecule design, combining chemical insights with machine learning prowess. Future directions might include further optimizations in computational efficiency, more nuanced understanding of chemical space through advanced GNNs, and the use of these tools in generative models for drug discovery.
MatterGen: A New Frontier:
- MatterGen represents a paradigm shift in materials design by employing generative AI to create new compounds with specific properties. Announced by Microsoft researchers, this model can design materials tailored to specific needs, such as efficient solar cells or CO2 recycling, advancing beyond traditional trial-and-error methods. MatterGen leverages the power of the previously discussed tools, integrating them into a system that can generate novel materials based on desired chemical, mechanical, electronic, or magnetic properties (5).
Microsoft researchers introduce MatterGen, a model that can discover new materials tailored to specific needs—like efficient solar cells or CO2 recycling—advancing progress beyond trial-and-error experiments. https://t.co/z9yOaV7VGo pic.twitter.com/qrmYHheKlS
— Microsoft Research (@MSFTResearch) January 16, 2025
This review underscores the synergy between chemical informatics and machine learning, where RDKit, PyTorch, TensorFlow, and now MatterGen collectively empower researchers to push the boundaries of molecular design, from academic research to practical applications in drug discovery and materials science.
My Journey:
I am currently on an exhilarating journey to develop a platform that will revolutionize how we design novel molecules, not only for materials science but also for therapeutic applications. By combining the capabilities of RDKit, PyTorch, TensorFlow, and MatterGen, I aim to create a system that can predict and design compounds with unprecedented precision and efficiency. To stay informed about my recent advancements, new codes, and the evolution of this project, I invite you to subscribe. Your support and interest will be integral to bringing these innovations from the digital realm into practical, real-world applications that could change the landscape of both materials science and medicine.

Author:
Navindra Soodoo, Ph.D. in Material Chemistry

